

Analytic Interatomic Forces in the Random Phase Approximation
Category: Scientific HighlightsPublished in Physical Review Letters
Introduction
In electronic structure theory, the possibility to calculate forces between the individual atoms is a crucial cornerstone for the broad usability of a method. If forces can be computed, structures can be relaxed and one can follow the motion of atoms at finite temperature. This opens the route to many applications that are otherwise not feasible. In the recent work of Ramberger and coworkers, a remarkably simple equation for the forces in the random phase approximation for the correlation energy is derived and tested. The method is not restricted to the random phase approximation, but can be applied to almost any diagrammatic many body method and is thus of general use for a wide class of theoretical methods. Considering that forces in solids were previously only computable within density functional theory, this work can be considered to be a major cornerstone in electronic structure theory.
Abstract
We discuss that in the random phase approximation (RPA) the first derivative of the energy with respect to the Green’s function is the self-energy in the GW approximation. This relationship allows us to derive compact equations for the RPA interatomic forces. We also show that position dependent overlap operators are elegantly incorporated in the present framework. The RPA force equations have been implemented in the projector augmented wave formalism, and we present illustrative applications, including ab initio molecular dynamics simulations, the calculation of phonon dispersion relations for diamond and graphite, as well as structural relaxations for water on boron nitride. The present derivation establishes a concise framework for forces within perturbative approaches and is also applicable to more involved approximations for the correlation energy.
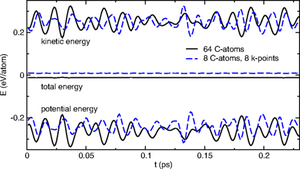
| For the first time, the RPA was used to perform a molecular dynamics simulation in solids. The figure shows the potential and kinetic energy per atom for diamond supercells of 64 and 8 atoms, using the Γ point and 2×2×2 k points, respectively. Energies are shifted so that the total energies approach zero. The slight fluctuations in the total energy are similar to those observed in DFT for a 1 fs time step using a Verlet algorithm. |
|---|
Project part: P02 Towards exact correlation in extended systems
Original publication: Ramberger, B., Schäfer, T., Kresse, G., 2017. Analytic Interatomic Forces in the Random Phase Approximation. Physical Review Letters 118. DOI:10.1103/PhysRevLett.118.106403
Published by the American Physical Society under the terms of the Creative Commons Attribution 4.0 International license.
Sensengasse 8/12
A-1090 Vienna
AUSTRIA
T: +43-1-4277-51401
F: +43-1-4277-9514